
Sňnia Orenga Montoliu
Diseny i presentació de treballs científics
“żQuién soy yo propiamente? Mi Respuesta es ésta: desde el instante en que tomé conciencia de mí, soy aquel en que libremente me he convertido y lo soy porque he sido yo quien me he convertido en él.”
Johann Gottlieb Fichte
De acuerdo con la evidencia disponible, la ECJ resulta del plegamiento anormal de un prión. Este fenómeno parece estimular a que otras proteínas alteren sus formas, afectando su capacidad para funcionar. Por esto, se la clasifica entre las enfermedades priónicas o encefalopatías espongiformes transmisibles (EET), caracterizadas por presentar una forma anómala de la proteína priónica celular (PrPC).El término espongiforme alude al aspecto esponjoso que presenta en la autopsia el cerebro afectado.
La enfermedad de Creutzfeldt-Jakob (ECJ) es un mal neurológico con formas genéticas hereditarias y también contagiosas, producidas por una proteína llamada prión (PrP). Si bien los casos hereditarios e infecciosos están perfectamente documentados, la causa de la aparición del prión es desconocida en la mayor parte de los casos informados.
Se trata de una enfermedad de naturaleza degenerativa y pronóstico mortal que afecta aproximadamente a una persona por millón (prevalencia de 1:106) a nivel global.
La enfermedad de Creutzfeldt-Jacob (ECJ) es una enfermedad degenerativa del SNC que está producida por unas proteínas infecciosas denominadas priones. Ocurre una demencia rápidamente progresiva y profunda, acompańada de ataxia cerebelosa, sacudidas mioclónicas difusas y otras diversas anomalías visuales y neurológicas. Los cambios neuropatológicos principales se producen en la corteza cerebral y cerebelosa, los aspectos sobresalientes son la pérdida neuronal generalizada y gliosis acompańada por vacuolación sobresaliente o estado esponjoso en las regiones afectadas (Adams R.; Victor M.; Ropper A., 2000, p.668), de aquí la denominación encefalopatía espongiforme subaguda (EES).
La Enfermedad de Creutzfeldt-Jakob (ECJ) pertenece a una familia de enfermedades de los seres humanos y animales conocidas como encefalopatías espongiformes transmisibles (EET). La ECJ es la más común de las encefalopatías espongiformes transmisibles (EET) humanas conocidas. Entre otras EET humanas figuran el kuru, el insomnio familiar fatal (IFF) y la enfermedad Gerstmann-Straussler-Scheinker (GSS).
Las primeras referencias a las enfermedades espongiformes transmisibles se remontan al siglo XVIII, cuando varios ganaderos europeos describieron una enfermedad neurodegenerativa letal que afectaba a las ovejas y a las cabras, mal al que se denominó "tembladera"; (en inglés, scrapie). El cerebro de estos animales presentaba un aspecto de esponja, de donde proviene el término "espongiforme". A principios del siglo XX se describieron los primeros casos de encefalopatía espongiforme en el ser humano, y la enfermedad se bautizó con el nombre de enfermedad de Creutzfeldt-Jakob. Posteriormente se demostró que estas enfermedades eran transmisibles.
El agente patógeno, el prión, fue descubierto en 1982 por Stanley Prusiner, quien demostró que se trataba de partículas puramente proteicas sin ácido nucleico. Prusiner y colaboradores presentaron pruebas convincentes de que el agente patógeno transmisible es una partícula proteinácea infecciosa desprovista de ácido nucléico, que resiste a la acción de las encimas que destruyen el RNA y DNA y que en las imágenes de microscopía electrónica no tiene la estructura de un virus (Adams R.; Victor M.; Ropper A., 2000, p. 668). Prusiner lo denominó prión. En 1997 le fue otorgado el Premio Nobel de Fisiología o Medicina.
Los priones o proteínas priónicas son partículas acelulares, patógenas y transmisibles. Se caracterizan por producir enfermedades que afectan el sistema nervioso central (SNC), denominadas encefalopatías espongiformes transmisibles (EET). Los priones no son seres vivos, a diferencia de los virus estos microorganismos son resistentes a un amplio abanico de tratamientos químicos y físicos (Murray P. et al., 2002, p.597), como el formaldehído, radiación ultravioleta y calor hasta 80şC.
El aislamiento de priones a través del seguimiento del nivel de infectividad en las EET demuestra que las partículas infectivas están constituidas total o parcialmente por una forma modificada de la proteína prión. El prión que carece de ácidos nucléicos detectables, consiste en agregados de glucoproteínas hidrófobas resistentes a la proteasa (Murray P. et al., 2002, p.597), denominados PrPSc. Se agrega en forma de bastón amiloide (fibrillas), se encuentra en vesículas endoplásmicas de la célula y se secreta. La proteína se expresa en varios tejidos, principalmente en neuronas del SNC, y se une a las membrana celular externa mediante una molécula de glicosil fosfatidil inositol (GPI).
Los resultados experimentales sugieren que la acción patógena de los priones está muy relacionada con la forma modificada de una proteína natural existente en el organismo que, al entrar en contacto con las proteínas originales, las induce a adoptar la forma anómala del prión, mediante un mecanismo todavía desconocido. Todo ello en una acción en cadena que acaba por destruir la operatividad de todas las proteínas sensibles.
 |
Modelos moleculares: estructuras de PrPC
|
La PrP es una proteína celular normal
de 30 kD presente en las neuronas. La enfermedad aparece cuando la proteína
criónica sufre una modificación en su configuración desde la isoforma en hélice
α normal: PrPC (c por "celular", ya que se halla presente en todas las células)
hasta una isoforma anormal con plegamiento β, que habitualmente se denomina
PrPSc (por “scrapie”). Al modificar su configuración, la proteína priónica
adquiere una resistencia relativa a la digestión con proteasas (Cotran R., 2000,
p. 1371), como la proteinasa K.
La proteína del prión (PrP), se encuentra codificada por un gen (PRNP), que se
encuentra sobre el brazo corto del cromosoma 20 (Adams R.; Victor M.; Ropper A.,
2000, p. 668). En los mamíferos los priones se reproducen uniéndose a la
isoforma celular normal de la proteína priónica (PrPc) y estimulando su
conversión en la isoforma que produce la enfermedad (PrPSc).
El estudio de los priones ha dado origen a cuatro nuevos conceptos. El primero es que los priones son los únicos agentes patógenos infecciosos conocidos que carecen de ácido nucleico. El segundo, que las enfermedades criónicas pueden manifestarse como trastornos infecciosos, genéticos o esporádicos (Prusiner S.; Bosque P., 2001, P.2910). Ningún otro grupo de enfermedades de etiología única presenta un espectro tan amplio de manifestaciones clínicas. El tercero, que las enfermedades priónicas se producen por la acumulación de PrPSc, cuya conformación difiere sustancialmente de la de su precursora PrPC. El cuarto y último, que la PrPSc puede existir bajo diferentes conformaciones, cada una de las cuales parece caracterizar a cada fenotipo específico de la enfermedad.
El gen productor del prión ha sido bautizado PRNP. Este gen se encuentra localizado en el brazo corto (p) del cromosoma 20, en la posición 20pter-p12 (es decir, entre la posición 20p12 y el final o término del brazo), ocupando 15.000 pares de bases: más precisamente, desde el par 4.615.068 al 4.630.233 del cromosoma. Codifica la proteína PrPSc, presenta un único exón[4] que codifica todo el marco de lectura abierto y muestra un elevado grado de conservación en las distintas especies. A pesar de esta conservación, las partículas infecciosas procedentes de una especie, son muy eficaces para transmitir la enfermedad a cualquier huésped (Cotran R., 2000, p. 1371) que contenga el gen de la proteína normal de esa especie.
En casos de ECJ de carácter familiar,
se ha identificado una amplia gama de mutaciones que causan la enfermedad. Por
ejemplo, ciertas familias con ECJ, la enfermedad está relacionada con una
mutación puntual (D178N) en el gen PRNP; además, en éste se han encontrado
varios polimorfismos. Se ha observado que el polimorfismo Met/Val en el codón
129 influye en el patrón de la enfermedad, la presencia de Val en el codón 129
causa la ECJ. En la forma esporádica de la ECJ se han observado otras
influencias del polimorfismo en el codón 129: muchos pacientes con esta
enfermedad son homocigotos en el codón [5]129 para Met o Val, mientras que casi
la mitad de los pacientes control son heterocigotos en esta localización (Cotran
R., 2000, p. 1371), lo que sugiere que la heterocigosidad en el codón 129 tiene
un carácter protector frente a la aparición de la enfermedad.
 |
Mapa del brazo corto del cromosoma 20.
|
La enfermedad de Creutzfeldt-Jakob se
puede agrupar en clásica o nueva variante. Los tipos clásicos de esta enfermedad
son:
ECJ de tipo esporádico (ECJe) que comprende la mayoría de los casos y que generalmente ocurre sin razones conocidas. La forma esporádica de la ECJ es la enfermedad priónica más frecuente (Prusiner S.; Bosque P., 2001, p. 2911) en seres humanos. Es responsable del 85% de todos los casos de enfermedad criónica humana.
El comienzo de una enfermedad
esporádica puede ocurrir, teóricamente, después de una mutación somática y
seguir, de este modo, una vía similar a la de las mutaciones de la línea
germinal en las formas hereditarias. Hasta la fecha se han identificado 20
mutaciones diferentes que provocan sustituciones no conservadoras en el gen PrP
humano (Prusiner S.; Bosque P., 2001, p. 2912) y que segregan con las
enfermedades criónicas humanas.
Resulta cuando una persona hereda la proteína anormal (prión). Este tipo hereditario es poco común, suponen del 10 al 15% de todos los casos. Mutaciones de sentido erróneo y expansiones en la región de repetición del octapéptido del gen son responsables de las formas familiares de la enfermedad priónica. Se han vinculado 5 mutaciones distintas del gen PrP a la enfermedad priónica hereditaria.
Aunque dentro de una misma familia los fenotipos pueden variar de forma llamativa, fenotipos específicos tienen a asociarse (Prusiner S.; Bosque P., 2001, p. 2912) con ciertas mutaciones.
El tipo clásico de la enfermedad de Creutzfeldt-Jakob no está relacionado con la enfermedad de las “vacas locas” (encefalitis espongiforme bovina).
Parece ser consecuencia de la inoculación accidental con priones de los pacientes. La transmisión accidental de la ECJ al hombre ha sido descrita en el transplante de córnea, la implantación de electrodos de electroencefalograma contaminados y en algunos procedimientos quirúrgicos (Prusiner S.; Bosque P., 2001, p. 2913) (antes de que se supiera cómo desinfectar adecuadamente los instrumentos).
Se han presentado algunos casos en adolescentes que han recibido hormona del crecimiento hecha de glándulas hipofisarias de cadáveres (cuerpos muertos). Los priones no pueden ser destruidos mediante las técnicas de desinfección ordinarias que se utilizan para evitar la transmisión de virus y bacterias. Como resultado, la hormona sigue estando contaminada.
La inoculación accidental de priones a algunos pacientes durante los procedimientos quirúrgicos parece deberse al empleo de algunos instrumentos o aparatos en el área quirúrgica contaminados (Prusiner S.; Bosque P., 2001, p. 2913) por haberse empleado al operar a algún paciente con ECJ. Aunque la epidemiología de estos casos es muy significativa, no hay pruebas que puedan confirmar su origen.
La restringida aparición geográfica y cronológica de la nueva variante de la ECJ ha suscitado la posibilidad de que los priones de la encefalopatía espongiforme bovina (EEB) se hayan transmitido a los seres humanos. En adolescentes y adultos jóvenes que ha aparecido en Europa, es el resultado de la exposición a la carne de vaca contaminada (Prusiner S.; Bosque P., 2001, p. 2911) procedente de ganado que padecía la encefalopatía espongiforme bovina (EEB) Es una forma infecciosa de CJD que está relacionada con la “enfermedad de las vacas locas”. Se cree que la infección responsable de la enfermedad de las vacas locas es la misma responsable de la nueva variante de CJD en humanos. La nueva variante de la enfermedad de Creutzfeldt-Jakob representa menos del 1% de los casos y tiende a afectar a personas más jóvenes.
Es posible que una determinada conformación de la PrPSc bovina se haya seleccionado por su resistencia al calor en el proceso de replicación y que haya sido seleccionada de nuevo muchas veces (Prusiner S.; Bosque P., 2001, p. 2913) cuando el ganado infectado por comer alimentos con carne y huesos es sacrificado y sus despojos convertidos en nuevos alimentos. Estudios de cerebros de pacientes que han muerto a causa de la nueva variante de ECJ han confirmado la existencia de un patrón de glucoformas de PrP cuando se intenta relacionar la EEB con la nueva variante de ECJ, por que la PrPSc se forma después de que la proteína sea glucosilada y la desglucosilación de la PrPSc requiere de su desnaturalización.
Los primeros estudios con ratones no transgénicos sugirieron que la nueva variante de ECJ y la EEB podrían tener un mismo origen (Prusiner S.; Bosque P., 2001, p. 2914), porque ambas fuentes de material infeccioso transmitían la enfermedad con períodos de incubación similares, aunque muy prolongados.
La enfermedad se manifiesta en todas las partes del mundo y en todas las estaciones, con una incidencia anual de uno a dos casos por millón de personas (Adams R.; Victor M.; Ropper A., 2000, p. 669). La incidencia informada es un poco más elevada en áreas urbanas que en las rurales.
La ocurrencia de casos familiares, sugiere susceptibilidad genética a la infección, aunque no puede despreciarse la posibilidad de exposición frecuente al agente transmisible. El único mecanismo claramente demostrable de diseminación es la EES yatrógena.
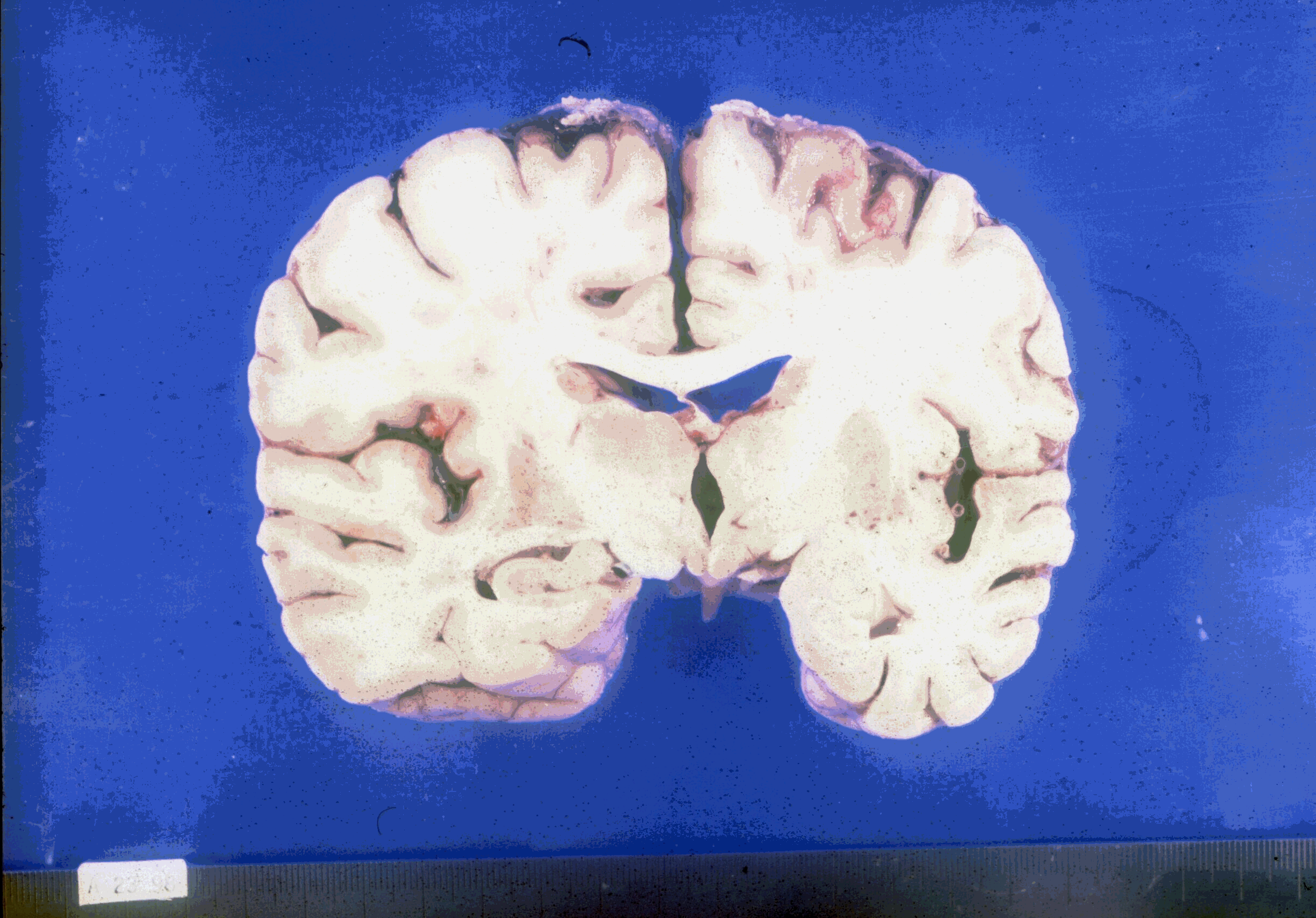
Con el microscopio óptico las características son la degeneración espongiforme y la astrogliosis. La encefalopatía espongiforme describe el aspecto de las neuronas vacuolizadas, así como su pérdida de función y la ausencia de reacción inmunitaria o inflamación. Se observa una aparición de vacuolas en las neuronas, formación de placas que contienen amiloide y fibrillas, proliferación e hipertrofia de los astrocitos y fusión de las neuronas (Murray P. et al., 2002, p.598) con las células adyacentes de la glía. Las neuronas y los fagotitos absorben la PrPSc, pero es difícil de degradar, una característica que puede contribuir a la vacuolización del tejido cerebral.
 |
Microfotografía de tejido encefálico en animal con EEB.
|
La degeneración y desaparición de células nerviosas se acompańa de proliferación astroglial intensa; estudios ultraestructurales han demostrado que las vacuolas microscópicas, que dan al tejido su aspecto esponjoso característico, se encuentran localizadas dentro de las proyecciones citoplásmicas de las células gliales y de las dendrítas de las células nerviosas. Esta transformación espongiforme consiste en un proceso multifocal que da lugar a la formación difusa de vacuolas pequeńas y aparentemente vacías, de diversos tamańos, en el neuropilo (entre los cuerpos neuronales, de numerosas vacuolas de 1 a 5 μm) y a veces en el pericarion de las neuronas.
La enfermedad afecta principalmente a las cortezas cerebral y cerebelosa, por lo general de manera difusa (Adams R.; Victor M.; Ropper A., 2000, p. 669), aunque en algunos casos están afectadas casi exclusivamente las regiones occipitoparietales. En resumen: corteza cerebral, el putamen, el núcleo caudado, el tálamo y la capa molecular del cerebelo. Se observa una proliferación difusa de los astrocitos fibrosos por toda la sustancia gris cerebral, los procesos de los astrocitos rellenos de filamentos gliales forman redes extensas.
Se han encontrado placas de amiloide en el 10% de todos los casos de ECJ. En la nueva variante de ECJ, vemos la presencia de “placas floridas”. Estas placas están formadas por un núcleo central de amiloide de PrP rodeado de vacuolas (Prusiner S.; Bosque P., 2001, p. 2914), con una distribución que recuerda a los pétalos de una flor.
Además, los priones alcanzan grandes concentraciones en el cerebro, lo que contribuye aun más a la destrucción tisular. Los priones también se pueden aislar en otro tipo de tejidos, pero únicamente causan lesión en el cerebro. No se genera ninguna reacción inflamatoria o respuesta inmunitaria ante el microorganismo (Murray P. et al., 2002, p.598), lo que distingue esta enfermedad de una encefalitis vírica clásica. En el líquido cefalorraquídeo de los individuos sintomáticos se puede detectar un marcador proteico (proteína cerebral 14-3-3).
La microscopía electrónica demuestra que las vacuolas son intracitoplásmicas y que están rodeadas por membrana. Se localizan en las prolongaciones neuronales y gliales. Podemos ver placas de kuru, son depósitos extracelulares de proteína anómala conglomerada (Cotran R., 2000, p. 1373), estas presentan positividad con la tinción de rojo Congo y con la de PAS. En los casos de variante de ECJ aparece de forma abundante en la corteza cerebral.
El cuadro clínico suele ser típico,
la mayoría de pacientes presenta un déficit en su función cortical, con
alteraciones iniciales sutiles en la memoria y la conducta. Unos pocos pacientes
presentan alteraciones visuales (variante de Heidenhain) o marcha atáxica
cerebelosa[6] (variante de Brownell-Oppenheimer) y descoordinación. Es habitual
que los déficits cerebelosos se sigan por una demencia rápidamente progresiva
asociada a menudo con contracciones musculares espásticas involuntarias (Cotran
R., 2000, p. 1373), e intensas tras una estimulación súbita (mioclonia[7] de
estimulación). Una pequeńa proporción de pacientes se manifiestan como ataxia.
Otros síntomas y signos consisten en disfunción extrapiramidal que se manifiesta
por rigidez, fascies de máscara o movimientos coreoateroides; signos
piramidales; crisis epilépticas, hipoestesia, parálisis supranuclear de la
mirada, atrofia óptica y signos vegetativos, como cambios en el peso corporal,
temperatura, sudación o la menstruación.
Una tercera parte, aproximadamente, de los pacientes con ECJ padece síntomas
prodrómicos inespecíficos (Prusiner S.; Bosque P., 2001, p. 2914), como fatiga,
trastornos de sueńo, pérdida de peso, cefalea, malestar y dolor mal definido.
En muchos pacientes el patrón EEG es distintivo, y cambia durante la evolución de la enfermedad desde lentificación difusa e inespecífica, hasta complejos estereotipados de ondas de alto voltaje (1 a 2 Hz) y complejos de ondas agudas sobre un fondo cada vez más lento y de bajo voltaje (Adams R.; Victor M.; Ropper A., 2000, p. 669).
La enfermedad es uniformemente mortal y tiene una duración promedio de sólo 7 meses, aunque algunos pacientes han sobrevivido durante varios ańos.
Con excepción de la biopsia cerebral, no existen pruebas específicas para la ECJ; si se ve el conjunto de alteraciones anatomopatológicas características de la ECJ, entonces el diagnóstico es razonablemente seguro. El diagnóstico necrópsico puede llevarse a cabo mediante el uso de antisueros frente a la PrP. Muchos estudios mediante transferencia Western[8] han demostrado la presencia de proteínas PrP inmunorreactivas que son resistentes a las proteinasas K en el cerebro de pacientes con ECJ (Prusiner S.; Bosque P., 2001, p. 2914). Se ha desarrollado una forma de inmunoanálisis cuantitativo y de gran sensibilidad basándose en los epítopos que están expuestos en la PrPc pero ocultos en la PrPSc.
Si el paciente tiene antecedentes familiares que sugieren una ECJ hereditaria, la secuenciación del gen PrP puede facilitar el diagnóstico.
TC: puede ser normal o mostrar atrofia cortical. RM: imágenes ponderadas de T2 o de difusión con un discreto aumento de la seńal de los ganglios basales (ni sensible ni específico). Análisis de LCR: casi siempre normal, pero puede contener un ligero aumento del número de proteínas.
ECG: suele ser útil en el diagnóstico de la ECJ. En la mayoría de casos avanzados aparecen descargas agudas repetitivas, de alto voltaje, trifásicas y polifásicas (Prusiner S.; Bosque P., 2001, p. 2914). Estas descargas, aunque no siempre, son simétricas y pueden tener mayor amplitud a un lado. A medida que la ECJ progresa, los ritmos normales de fondo se fragmentan y se vuelven más lentos.
No se conoce ningún tratamiento eficaz para evitar o prevenir la ECJ. No existe actualmente tratamiento que pueda curar, mejorar ni siquiera controlar la sintomatología en la enfermedad de Creutzfeldt-Jakob. Los investigadores han sometido a prueba muchos fármacos, entre ellos la amantadina, los esteroides, el interferón, el aciclovir, la clorpromazina, y diversos agentes antivirales y antibióticos. No obstante, ninguno de estos tratamientos ha demostrado ser beneficioso.
El único tratamiento posible de la ECJ tiene como propósito principal aliviar los síntomas hasta donde sea posible y mejorar la calidad de vida del paciente.
El diseńo de fármacos atendiendo a su estructura y en relación con la inhibición negativa dominante de la formación de priones ha dado lugar a varios productos prometedores.
La prevención más segura es seguir una serie de normas higiénicas, sobre todo precauciones durante la asistencia médica y la manipulación de los materiales de pacientes con ECJ.
Para su eliminación se puede recurrir a un proceso de autoclave[9] a 15 psi durante una hora, tratamiento con una solución de hipoclorito al 5% o de hidróxido sódico 1’0M (Murray P. et al., 2002, p.600) puesto que estos agentes se pueden transmitir con los instrumentos y electrodos cerebrales, estos objetos se deben desinfectar cuidadosamente antes de volver a utilizarlos.
Adams, R.; Victor, M.; Romper, A. 2000 Principios de neurología
México: McGraw Hill. Interamericana
Bermejo F., Sáiz R., Díaz J. (2003)
Demencia vascular y otras enfermedades que cursan con demencia (on line)
Ediciones Doyma
Disponible en: http://db.doyma.es/cgi-bin/wdbcgi.exe/doyma/mrevista.go_fulltext_o_resumen?esadmin=si&pident=13046975
Cantor, D. (2007)
Medlineplus enciclopedia médica: enfermedad de Creutzfeldt Jacob (on line)
Adam
Disponible en: http://www.nlm.nih.gov/medlineplus/spanish/ency/article/000788.htm
21 Congreso de la SEAP, Madrid (2003) (On line)
SEAP. Sociedad Espańola de Anatomía Patológica
Disponible en: http://www.conganat.org/seap/congresos/2003/cursoautopsia/Image271.gif
Cotran, R. 2000 Patología Estructural y Funcional
Madrid: McGraw Hill. Interamericana
La Enfermedad de Creutzfeldt-Jakob: National Institute of Neurological Disorders
and Stroke (NINDS) (2008) (on line)
National Institute of Neurological Disorders and Stroke (NINDS)
Disponible en: http://espanol.ninds.nih.gov/trastornos/laenfermedad_de_creutzfeldt_jakob.htm
Enfermedad de Creutzfeldt-Jakob, Wikipedia, la enciclopedia libre (2008) (on
line)
Wikimedia Foundation, Inc.
Disponible en: http://es.wikipedia.org/wiki/Enfermedad_de_Creutzfeldt-Jakob
Murray, P. et al. 2002 Microbiología Médica
Madrid: Mosby. Elsevier Science
PRNP: prion protein (p27-30) (Creutzfeldt-Jakob disease, Gerstmann-Strausler-Scheinker
syndrome, fatal familial insomnia. (2008) (on line)
Genetics home referente: your guide to underestanding genetic conditions. U.S.
National Library of Medicine
Disponible en: http://ghr.nlm.nih.gov/gene=prnp
Prusiner, S.; Bosque, P. 2001 Enfermedades por Priones p.2910-2915
Kasper D. et al (Ed.) Harrison. Principios de Medicina Interna
Madrid: McGraw Hill. Interamericana

Actualizada el 3/06/08
{kind=link}